医薬品に関わる開発・申請支援サービス

医薬品のCMC薬事の専門家が、申請業務の煩雑さを解消し、迅速かつ確実な申請成功へ導きます。
サービス内容
開発段階から承認申請、申請後のフォローまで、品質/CMCに関する薬事業務の全てをワンストップで支援します。
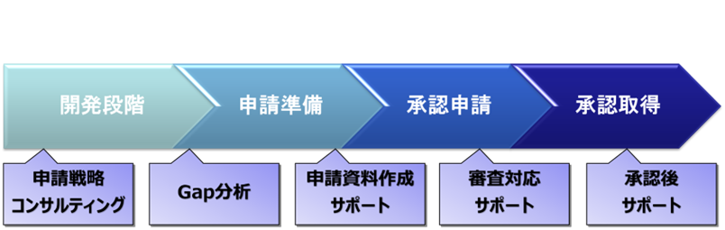

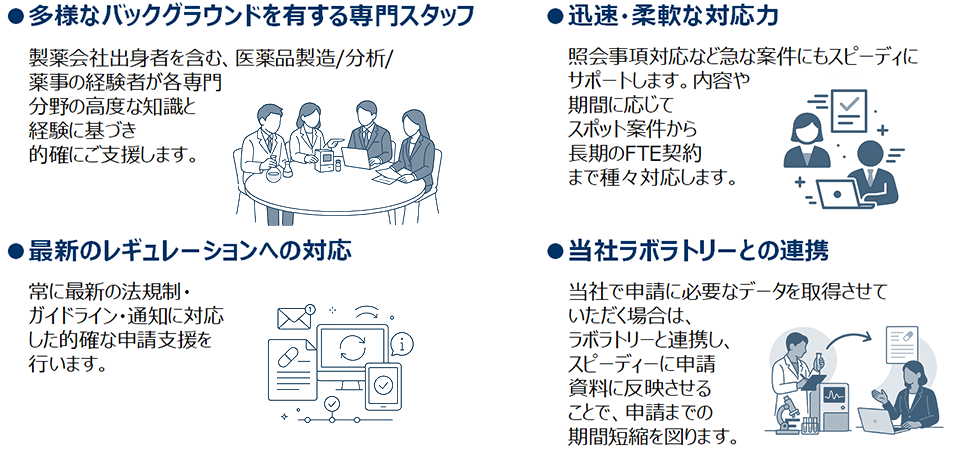
当社の強み
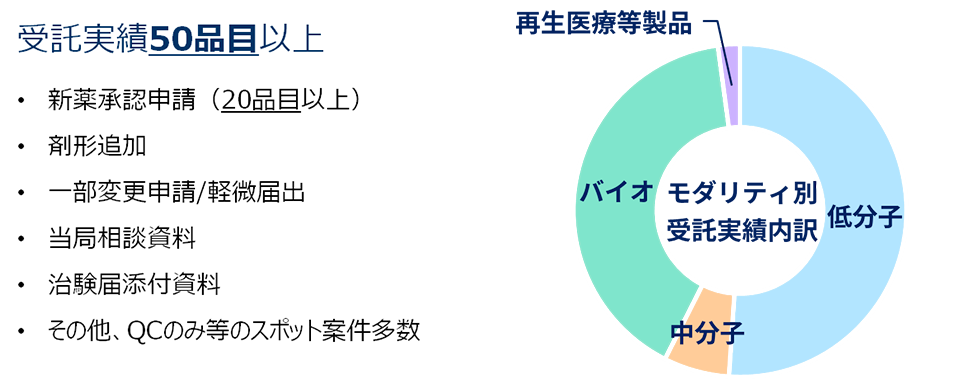
実績
よくあるご質問
Q.申請に必要な準備期間はどれくらいですか?
A. 新薬申請の場合は概ね1年、一部変更申請や剤型追加の場合は概ね6ヵ月ですが、申請内容や対象薬、データの充足度によって異なるため、ヒアリングを元に最短、最適なプランをご提示します。
Q.自社で経験のないモダリティーの申請を行いたい。
A.低分子だけでなく、ペプチド、抗体、血液製剤、ワクチン、再生医療等製品など、種々のモダリティーの新薬の申請をご支援させていただいた経験がありますので、どのようなモダリティーの製品もお任せください。また、医療機器とのコンビネーション製品にも対応した経験がございます。
Q.海外導入品の申請を速やかに進めたい。
A.海外の申請資料から国内向けの日本語のCTD第2部や承認申請書の作成をご支援した経験が多数ございますので、日本申請に向けたGAP分析から対応させていただくことも可能です。また、ご要望があれば、導入元企業様と直接やり取りして申請業務を進めることも可能です。
Q.分析も合わせて委託したいが可能か。
A.もちろん可能です。申請用データ取得や上市後の出荷試験も実施いたします。申請資料の作成や照会事項対応と併せてご依頼いただければ、ワンストップでスピーディーに対応させていただくことが可能です。
Q.QCだけ依頼することも可能か。
A.マスターバッチレコード(MBR)やSOPとの整合性確認を含め、専門的な観点でチェックさせていただきます。
Q.具体的な相談の前に料金の目安を知りたいです。
A.申請内容や資料のボリューム、ご希望されるサービス内容によるため、まずはヒアリングをさせていただいた上でお見積りをご提示しています。まずはお問い合わせフォームよりご連絡ください。
お問い合わせ・ご相談
